How cystic fibrosis promotes lung infections
From the NIH
At a Glance
- Scientists discovered how cystic fibrosis raises the risk for lung infections.
- The findings also identify a novel potential therapeutic target.
Cystic fibrosis is an inherited disorder that results in a buildup of thick and sticky mucus in the lungs, airways, and other organs. Excess mucus in the lungs can lead to coughing, breathing problems, scarring (fibrosis), and an increased risk of lung infections. The disorder affects about 30,000 people in the U.S. and 70,000 worldwide. There is no cure, but treatments can improve both the length and quality of life for people with the disease.
Cystic fibrosis is caused by mutations in the gene that encodes the cystic fibrosis transmembrane conductance regulator (CFTR). This protein functions as a channel across the membrane of cells that produce mucus, sweat, saliva, tears, and digestive enzymes. After the discovery linking the gene to the disease, scientists produced mice with a disrupted Cftr gene (CF mice). Surprisingly, the animals’ airways didn’t develop the bacterial infections typical of cystic fibrosis. The reasons why were a mystery.
To gain insights into the molecular basis for respiratory infections in people with cystic fibrosis, an NIH-funded group at the University of Iowa examined why CF mice don’t develop airway infections. The investigation, led by Viral Shah and Dr. Michael Welsh, was published on January 29, 2016, in Science.
One function of CFTR is to secrete bicarbonate, which has a high pH, into the liquid on the surface of airways, where respiratory infections begin. Loss of this activity leads to a more acidic airway surface in humans. Thus, the team first compared differences between the pH of airway surface liquid in mice and humans. They also studied CF pigs, which develop airway disease similar to humans with cystic fibrosis. Experiments suggested that pigs and humans have a mechanism to acidify airway surface liquid that is either absent or less efficient in mice.
The scientists assessed several candidate transporters to determine which makes pig airways acidic. Gene expression levels for ATP12A were abundant in the airways of pigs and humans, but very low in mouse airways. In several sets of experiments, the team showed that ATP12A activity is likely responsible for creating an acid environment that impairs airway defense in pigs and humans with cystic fibrosis.
Further experiments in CF pigs showed that inhibiting ATP12A raised airway surface liquid pH, enhanced bacterial killing, and reduced airway surface liquid viscosity (made it less thick and sticky). Conversely, raising levels of ATP12A in the airways of CF mice made the airway surface more acidic and impaired bacterial killing. The lungs of CF mice that expressed ATP12A had 100 times as many bacteria as those of control mice.
Taken together, the results show that CFTR, which secretes bicarbonate, and ATP12A, which secretes acid, balance each other to control airway surface pH. The loss of CFTR leaves acid secretion unchecked so that the airway surface becomes more acidic, thicker, and more hospitable for bacteria.
“This discovery helps us understand the cause of lung disease in people with cystic fibrosis. It may also identify ATP12A as a new therapeutic target,” Shah says. “We wonder if blocking ATP12A in people with cystic fibrosis could halt the progression of lung disease.”
—by Harrison Wein, Ph.D.
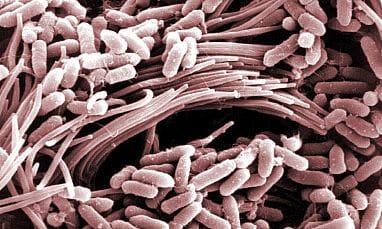
The Featured Image:
Scanning electron micrograph shows bacteria (Pseudomonas aeruginosa) on the surface of a CF-affected airway. Tom Moninger, University of Iowa Health Care
Related Links
- Cystic Fibrosis Therapy Tested in Young Children
- A Breakdown in Breathing: The Complexities of Cystic Fibrosis
- Cystic Fibrosis
- Cystic fibrosis (Genetics Home Reference)
Reference:
Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Shah VS, Meyerholz DK, Tang XX, Reznikov L, Abou Alaiwa M, Ernst SE, Karp PH, Wohlford-Lenane CL, Heilmann KP, Leidinger MR, Allen PD, Zabner J, McCray PB Jr, Ostedgaard LS, Stoltz DA, Randak CO, Welsh MJ. Science. 2016 Jan 29;351(6272):503-7. doi: 10.1126/science.aad5589. PMID: 26823428.


