New Guidelines for Cystic Fibrosis Diagnosis

New Guidelines for Cystic Fibrosis Diagnosis
A group of 32 scientists from nine countries have updated the guidelines for Cystic Fibrosis diagnosis. They published an article last month in The Journal of Pediatrics and the synopsis can be found here in Science Daily.
The scientists looked at data from people with Cystic Fibrosis worldwide to categorize hundreds of mutations, to better define the likelihood that someone with a mutation will have the disease and to better personalize treatment.
Called CFTR2 (the Clinical and Functional TRanslation of CFTR), the project began in 2008 and has thus far described about 300 out of the 2,000 known mutations, making it the most comprehensive compilation and evaluation of disease liability for all genetic diseases. As a result of CFTR2, mutations are now categorized as either cystic fibrosis causing, mutations of varying clinical consequence, non-cystic fibrosis causing or unknown.
Also, of major significance, the new guidelines lower the threshold for sweat tests. These are the previous guidelines:
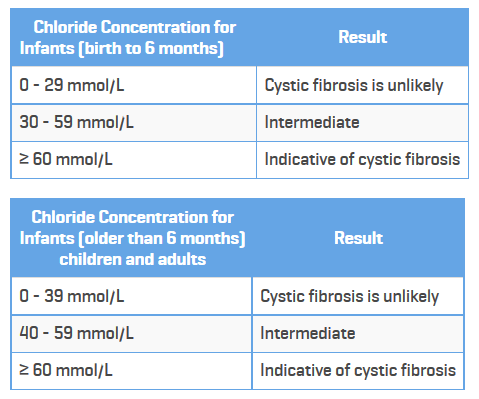
Here is information on the new guidelines, which change the borderline or “possible” category to above 30 mmol for all ages.
Based on data collected from CFTR2 and other research recognizing all cases of cystic fibrosis, the new guidelines lowered the threshold for “possible” cystic fibrosis from 40 millimoles per liter to 30 millimoles per liter for all ages. A normal range for chloride concentration in sweat is 10 to 20 millimoles per liter, and 60 millimoles per liter constitutes a cystic fibrosis diagnosis. A result of this updated guideline is that those with a chloride level between 30 and 40 millimoles per liter who were previously considered unlikely to have cystic fibrosis will now be reconsidered as possibly having cystic fibrosis or a related disease.
To learn more about the CFTR2 project, go to the website here.


